|
Three years ago I started my PhD studies on the subject of evolutionary biology. Three years in which I haven't stop learning new things and acquired new skills, mainly in the field of genomics and bioinformatics. Three years in which I have been able to attend to four international workshops and one big evolution conference, as well as many in-house seminars. Three years in which I have been able to visit six countries and I met a lot of interesting people. Three years in which I had mostly positive experiences. Three years that would have not been possible without the advices of my supervisor and many other collaborators. Three years that would have been impossible if I had listened to those who I did not listen. 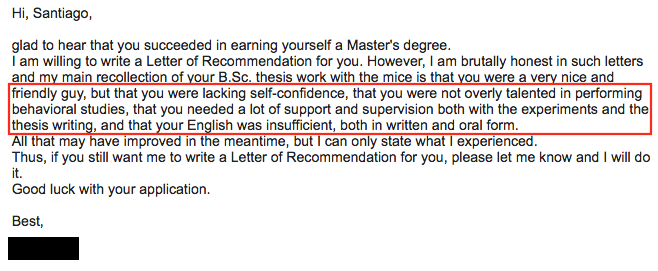 But most importantly, three years in which I did never give up. Above is a super harsh email that a Professor wrote to me when asking him/her for a recommendation letter in order to apply for my current PhD position. I replied kindly and rejected his/her recommendation letter. Three years later, I am self-confident enough to let everyone know how unfair this senior Professor was to me, which by that time I was just an inexperienced student - and as so, of course I needed some support. However, it still remains unclear to me if a "a lot of support and supervision" was referring to the only day that this person was present in the experiment room with me or maybe to the two revisions that he/she did to my manuscript. Anyway, three years.
5 Comments
No science today. Here just to let you know that after a long time as a "task" in my to-do list, I have now updated my Flickr page with the most amazing photos that I have taken during the last two years. Of course, I expect my gallery to keep growing as time / life moves on. But for the time being, here I share with you what I believe is my top 3 favorite photos:
#1: Porto, Portugal
#2: Barcelona, Spain
#3 Uppsala, Sweden
And that's all for today. I am sorry (to myself) for not posting more often. Anyway, I guess that is also kind of good because it means that I am quite busy recently. Note: If you are interested in seeing some more photos, just go to my www.flickr.com/people/santiago-montero-mendieta
Welcome to a new section in my blog that I will name "Did you know...?". Every once in a while, I will try to give a brief answer to two science-related questions. I will keep it very simple since my aim is simple too: I just want to gain (and share) some knowledge on popular issues. Let's start:
I am happy to confirm that I am going back to Czech Republic at the end of this month to a new workshop ☺ This time I am enrolled in the 2018 Workshop on Population and Speciation Genomics.
This workshop takes place from 21 January - 3 February (❄ ❄ ❄) at the UNESCO World Heritage Site Český Krumlov and it is organized by some of the best faculty members in the field of population and speciation genomics, such as Walter Salzburger, Michael Matschiner (University of Basel), David Swofford (Duke University) or Chris Jiggins (University of Cambridge) among many others. Interestingly, I already met David Swofford in last year's Workshop on Phylogenomics; plus I also attended a keynote given by Chris Jiggins in the 2017 Congress of the European Society for Evolutionary Biology. By participating in this workshop I expect to become familiar with the use of genomic data to study the evolutionary history on the level of populations and closely related species. Even though during the two previous years of my PhD thesis I have definitely gained experienced in various sorts of genomics analyses, I think this workshop is a very good opportunity for me to understand demography inference approaches in detail, learning from some of the leading experts in the field. This two-week workshop will be the fifth one (and maybe the last one) in which I will participate during my PhD studies. Český Krumlov is a charming fairy tail town; the perfect place for a science workshop. The food is super-tasty (not expensive at all) and hotel facilities are clean and comfortable. I am so excited to go back. Here some photos that I took last year: First of all, let me say start by saying: Happy New Year 2018! ☺
Interesting facts: Did you know that the number 2018 has only 4 divisors? These are the numbers 1, 2, 1009 and 2018. Interestingly, the sum of all these numbers gives us 3030. I can't really imagine how the world will look like by the year 3030, can you? In 2018 we also commemorate the centenary of the end of the First World War (1914-1918), in which the Allies (France, Britain, and Russia) defeat Germany and Austria-Hungary (the Central Powers). The year 2017 was an amazing time for learning new things, meeting new people and traveling to new places. In particular, during 2017 I established a new personal record, since I stayed in 4 different countries (Czech Republic, Sweden, The Netherlands and Spain). Scientifically speaking, 2017 was also a very fruitful year for me with 3 new publications, and 11 new citations. I attended a very interesting Workshop on Phylogenomics and also my first international conference (the 2017 Congress of the European Society for Evolutionary Biology) in which I obtained the Second Best Poster Award. Will the 2018 be a (even) better year? Time will reveal. Here is my 2018 new year resolution: to write a new entry into this blog at least once a month. In bioinformatics, many times I need to retrieve information from very large databases (e.g. 1,000,000 rows and 10 columns) for a target list of records (e.g. 1,000 IDs within the database). For example, suppose that we have a database of 1,000,000 SNPs with information on which gene each SNPs sits in. But we just need to know the genes for a target list of 1,000 SNPs within the database. This is a common task that I never do manually since there is a big chance of introducing errors. We need to write some code. I always use Perl, but I believe this can be solved using any programming language (e.g. Python, R...). In Perl, this can be solved very easily by using a HASH table. Here is my code to do that: As you can see, this is a very basic use of Perl scripting to solve a fundamental task in bioinformatics. Feel free to use this script or share it with someone that might need it. That's all for today!
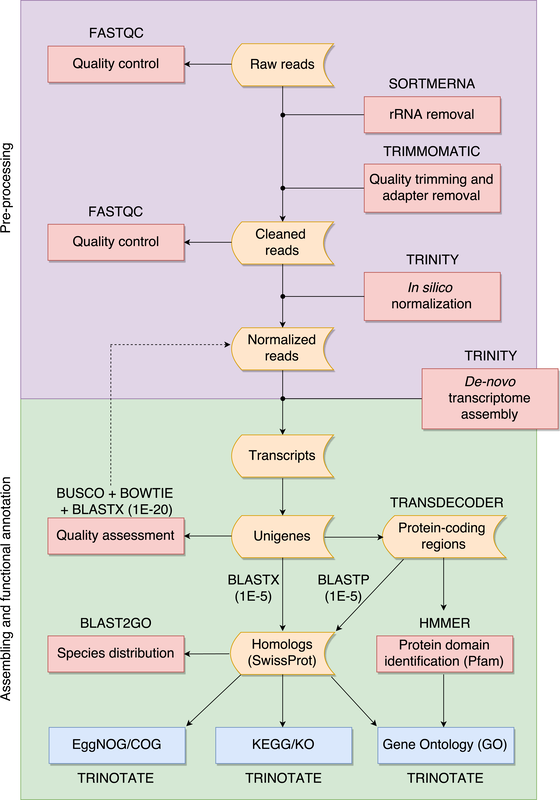 Whole genome sequencing (WGS) is a very valuable resource to understand the evo- lutionary history of poorly known species. However, in organisms with large genomes, as most amphibians, WGS is still excessively challenging and transcriptome sequencing (RNA-seq) represents a cost-effective tool to explore genome-wide variability. Non- model organisms do not usually have a reference genome and the transcriptome must be assembled de-novo. We used RNA-seq to obtain the transcriptomic profile for Oreobates cruralis, a poorly known South American direct-developing frog. In total, 550,871 transcripts were assembled, corresponding to 422,999 putative genes. Of those, we identified 23,500, 37,349, 38,120 and 45,885 genes present in the Pfam, EggNOG, KEGG and GO databases, respectively. Interestingly, our results suggested that genes related to immune system and defense mechanisms are abundant in the transcriptome of O. cruralis. We also present a pipeline to assist with pre-processing, assembling, evaluating and functionally annotating a de-novo transcriptome from RNA-seq data of non-model organisms. Our pipeline guides the inexperienced user in an intuitive way through all the necessary steps to build de-novo transcriptome assemblies using readily available software and is freely available at:
https://github.com/biomendi/TRANSCRIPTOME- ASSEMBLY-PIPELINE/wiki Full reference: Montero-Mendieta S, Grabherr M, Lantz H, De la Riva I, Leonard JA, Webster MT, Vilà C. (2017) A practical guide to build de-novo assemblies for single tissues of non-model organisms: the example of a Neotropical frog. PeerJ 5, e3702 [PDF] 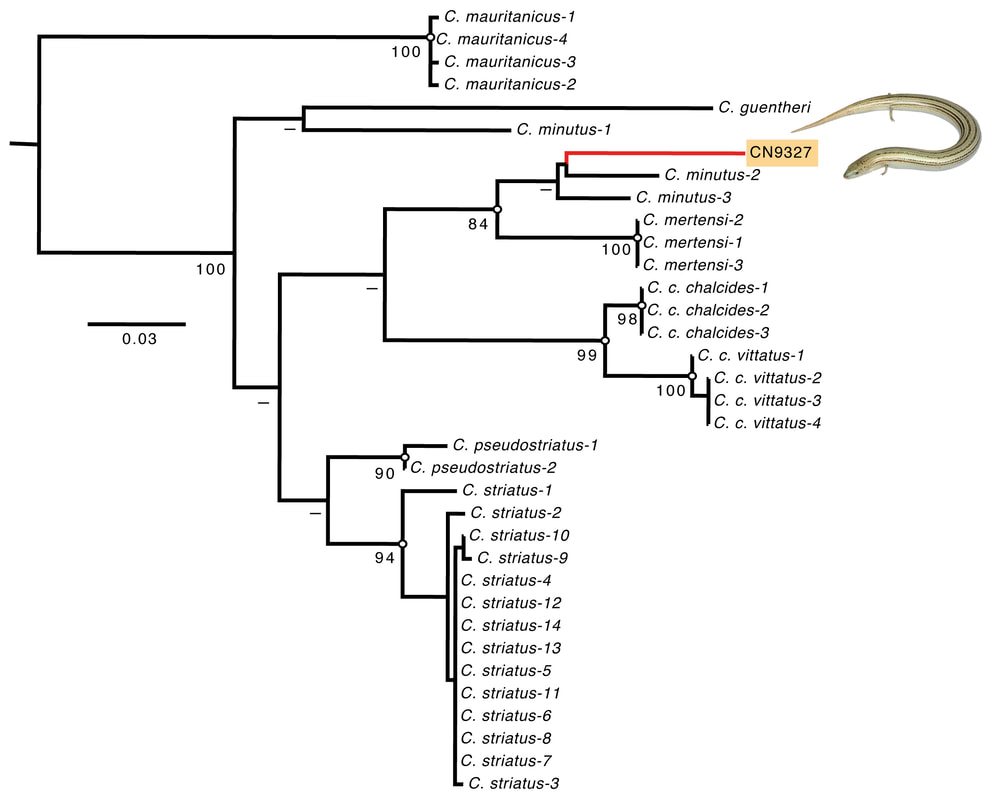 The genus Chalcides comprises about 30 species of scincid lizards mainly distributed across north Africa, its taxonomic status and distribution as described in the literature has fluctuated in recent years. in May 2014, the authors found a skink of the Chalcides type in Théniet el Had national Park (Algeria) initially classified as Chalcides mertensi Klausewitz, 1954, based on its morphological similarity and distribution. A region of 396-bp of the cytochrome b mitochondrial gene was sequenced as a reference against a preexisting phylogeny of the genus Chalcides. Surprisingly, this skink was genetically closely related with specimens of Chalcides minutus Caputo, 1993, found 300 km away. Comparison of the morphology between the new record and the original descriptions showed that this skink is likely to represent a new species of Chalcides, and that a major revision of Algerian skinks is needed to unravel the phylogeny of the C. minutus-mertensi species complex.
Full reference: Montero-Mendieta S, Ferrer J, Hammou MA, Dahmani W, Sanuy D & Camarasa S (2017) Another record or a new taxon? A candidate species of Chalcides Laurenti, 1768, in North Africa (Squamata: Sauria: Scincidae). Herpetozoa. Volume 29 (3/4): 155-161[PDF]  We studied the variation in genetics, bioacustics, and morphology in Eleutherodactylus glamyrus, a regionally endemic frog species restricted to high elevations in the Sierra Maestra Massif, Western Cuba that was originally described as a cryptic species hidden under the name E. auriculatus. Genetic analysis of mtDNA sequences of the 16S and cob genes identify two allopatric and strongly supported mitochondrial clades (phylogroups) which also showed no haplotype sharing in the nuclear Rag-1 gene. Bioacustic, and morphological comparisons concordantly identify these two phylogroups as independent evolutionary lineages. Therefore, we herein restrict the name Eleutherodactylus glamyrus Estrada and Hedges to populations represented in our analyses as the western phylogroup (Cordillera del Turquino to Pico La Bayamesa) and consider specimens from the eastern phylogroup (Sierra del Cobre) to represent a new species described and named as Eleutherodactylus cattus. Our results add to the growing list of Eleutherodactylus species endemic to Cuba and highlight the importance of combining different sources of evidence for obtaining robust assessments of species limits in amphibians.
Full reference: Rodríguez A, Dugo-Cota A, Montero-Mendieta S, Alonso R, Vences M, Vilà C. (2017) Cryptic within cryptic: genetics, morphometrics, and bioacoustics delimitate a new species of Eleutherodactylus (Anura: Eleutherodactylidae) from Eastern Cuba. Zootaxa 4221 (5): 501–522 [PDF] A decade ago, DNA barcoding was proposed as a fast, cost-efficient and simple taxonomic method based on the use of a unique, short and standardized gene region for identifying existing species and speeding the discovery of new ones. Few DNA barcoding studies of squamate reptiles have been conducted. Due to the significance of the Socotra Archipelago (a UNESCO Natural World Heritage site and a biodiversity hotspot) and the conservation interest of its reptile fauna (94% endemics), we performed the most comprehensive DNA barcoding study on an island group to date to test its applicability to specimen identification and species discovery. Reptiles constitute Socotra’s most important vertebrate fauna, yet their taxonomy remains under-studied. We successfully DNA-barcoded 380 individuals of all 31 presently recognized species. The specimen identification success rate is moderate to high, and almost all species presented local barcoding gaps. The unexpected high levels of intra-specific variability found within some species suggest cryptic diversity. Species richness may be under-estimated by 13.8–54.4%. This has implications in the species’ ranges and conservation status that should be considered for conservation planning. Other phylogenetic studies using mitochondrial and nuclear markers are congruent with our results. We conclude that, despite its reduced length (663 base pairs), cytochrome c oxidase 1, COI, is very useful for specimen identification and for detecting intra-specific diversity, and has a good phylogenetic signal. We recommend DNA barcoding to be applied to other biodiversity hotspots for quickly and cost-efficiently flagging species discovery, preferentially incorporated into an integrative taxonomic framework.
Full reference: Vasconcelos R*, Montero-Mendieta S*, Simó-Riudalbas M, Sindaco R, Santos X, Fasola M, Llorente GA, Razzetti E, Carranza S. (2016) Unexpectedly high levels of cryptic diversity uncovered by a complete DNA barcoding of reptiles of the Socotra Archipelago. PLOS ONE 11(3): e0149985 (*Both authors contributed equally to this work) [PDF] [Press release] [IBOL press]  On 13th June 2014 we observed one population of Alpine newt (Ichthyosaura alpestris) outside its natural distribution range, near the natural space of "Parc del Castell de Montesquiu" in Barcelona, Catalonia Spain. This finding represents the first report of a breeding population of I. alpestris in Catalonia. On 18th and 25th June, we performed two samplings with the aim of: a) determining whether nearby water ponds were colonized, b) estimating population size, c) performing population biometrics, d) determining the origin of the introduced specimens, and e) detecting the presence of pathogens in the population. After doing molecular analyses, we found out that the introduced might be a mix of individuals from two different subspecies: I. a. apuanus and I. a. cyreni. Interestingly, these subspecies are commonly used in aquariums. We detected the presence of Ranavirus spp in the studied individuals, thus increasing the risk of transmission to native species, such as Alytes obstetricans. Additionally, we also observed the presence of two other breeding populations of I. alpestris near the study area, which might be a direct consequence of their invasive potential. We concluded that a drastic intervention is highly needed to prevent the expansion of I. alpestris and remove the species from this area.
Full reference: Fibla M, Ubach A, Oromí N, Montero-Mendieta S, Camarasa S, Pascual-Pons M, Martínez-Silvestre A & Montori A. (2015) Población introducida de tritón alpino (Mesotriton alpestris) en el Prepirineo catalán. Bol. Asoc. Herpetol. Esp: 26(1) [PDF] |

Hi, I am Santi. This blog series was mainly created to include a summary of each of my publications. However, this blog is also a place where I will write about science and my life as researcher in the field of evolutionary biology.
Categories
All
|
